Table of Contents
Definition
noun
plural: glycogens
gly·co·gen, glī′kə-jən
A multi-branched polymer of glucose, mainly produced in liver and muscle cells, and functions as secondary long-term energy storage in animal cells
Details
Overview
Glycogen belongs to a group of polysaccharide carbohydrates. Carbohydrates are organic compounds comprised of carbon, hydrogen, and oxygen, usually in the ratio of 1:2:1. They are one of the major classes of biomolecules. As a nutrient, they can be classified into two major groups: simple carbohydrates and complex carbohydrates. Simple carbohydrates, sometimes referred to as simply sugar, consist of one or two saccharide residues. They are readily digested. They serve as a rapid source of energy. Complex carbohydrates (such as cellulose, starch, chitin, and glycogen) are those that need more time to be digested and metabolized. They are often high in fiber and unlike simple carbohydrates they are less likely to cause spikes in blood sugar levels.
History and terminology
Claude Bernard 1813 –1878, a French physiologist, was credited as the one who discovered glycogen. He was the first to describe how he isolated a substance from the liver and its properties. He called the substance la matière glycogène (“sugar-forming substance”) in 1857.1 The chemical formula of glycogen, (C6H10O5)n was established by the German organic chemist Friedrich August Kekulé 1829-1896 in 1858.
Characteristics
In animals, the glycogen is one of the main forms of energy reserves (the other being triglycerides or body fat). Similar to starch, the glycogen is a complex carbohydrate that stores excess glucose. It is sometimes referred to as “animal starch.” That is because the amylopectin constituent of plant starch is similar in composition and structure with the polysaccharide constituent of glycogen. The difference is the extensive branching in glycogen at every 8 to 12 glucose units.
The glucose units are linked by α(1→4) glycosidic bonds, forming a chain. The branches are linked to the glucose chains by α(1→6) glycosidic bonds. α-Glycosidic bonds form open helical polymers (as opposed to β-glycosidic bonds that produce nearly straight strands forming structural fibrils like in cellulose).2 Under the microscope, glycogen has a characteristic asterisk or star appearance. It occurs as a granule in the cytosol of the cell. The diameter ranges from 10 to 40 nm.2 At the core of a glycogen granule, there is a glycogenin, which is the enzyme that catalyzes the conversion of glucose to glycogen and serves as a primer.
Biological reactions
Biological reactions
The chemical process of joining monosaccharide units is referred to as dehydration synthesis since it results in the release of water as a byproduct. Starch is produced by dehydration synthesis, particularly by displacing a hydroxyl radical from one glucose and a proton from another glucose, and then, linking the two by a glycosidic bond. The detached hydroxyl radical and proton (hydrogen ion), in turn, join and form a water molecule. Glycogen is synthesized naturally in the animal body, particularly in the cells of the liver and skeletal muscles. Small amounts of glycogen can be found in the kidneys, some glial cells in the brain, and the white blood cells. The uterus also stores glycogen during pregnancy to nourish the embryo. The process of biosynthesizing glycogen from glucose is called glycogenesis.
Glycogenesis is the metabolic process of producing glycogen from glucose for storage in response to the high glucose level in the bloodstream. In humans and other animals, the two major sites of glycogenesis are the liver cells and the skeletal muscle cells. In the liver, there is about 10% glucose by weight whereas 2% glucose in skeletal muscle. (Overall, though, there are more glucose in the skeletal muscle because the latter’s mass is greater than the liver).2 In the liver cell, glucose is phosphorylated by glucokinase at position 6, thus producing glucose 6-phosphate. The phosphorylation of glucose traps it inside the cell. In other cells, glucose enters passively, and then, it is phosphorylated through hexokinase. This results in a compound that cannot leave the cell as well. Short polymers of glucose, especially exogenous glucose, are converted into long polymers to be stored inside the cell. The process is reversible though. When the body requires metabolic energy, glycogen is broken down into glucose subunits through the process of glycogenolysis.
As noted earlier, each glycogen granule has a glycogenin. It is a glycosyltransferase enzyme that primes the polymerization of glycogen. Before the main enzyme glycogen synthase can catalyze the addition of glucose, there should be an initial chain of at least three glucose residues. This is enabled by the catalytic activity of glycogenin, which, in this case, serves as a primer. Glycogenin catalyzes the transfer of glucose residues from UDP-glucose to itself by forming the α-1,4-glycosidic bond in order to create a polymer of glucose. Only when there are enough glucose residues in a chain that glycogen synthase will act, i.e. by extending the chain.
Biological reactions
When the body needs energy, glycogen is broken down into glucose with glucagon. Glycogenolysis is the process of breaking down stored glycogen in the liver so that glucose may be produced for use in energy metabolism. Thus, glycogenolysis is somewhat the opposite process of glycogenesis. Stored glycogen in the liver cells is broken down into glucose precursors. A single glucose molecule is cut off from the glycogen and is converted into glucose 1-phosphate, which in turn, is transformed into glucose 6-phosphate. Glucose 6-phosphate has three fates: (1) enter glycolysis, (2) processed by pentose phosphoate pathway, and (3) conversion into free glucose to regulate and maintain blood glucose levels. (In plants, the process of breaking down stored starch is called starch degradation)
Biological reactions
Hydrolysis is the process of converting polysaccharide into simple monosaccharide components. The process of converting polysaccharides into monosaccharides, in particular, is called saccharification. The water molecule is used in the process. In humans, carbohydrates (apart from monosaccharides) are digested through a series of enzymatic reactions. These enzymes are salivary amylase, pancreatic amylase, and maltase. In the mouth, complex carbohydrates such as glycogen and starch are initially broken down by the enzyme salivary amylase. The bolus travels though the gastrointestinal tract. The acidic gastric content of the stomach inhibits the digestive activity of the salivary amylase. Thus, the next phase of carbohydrate digestion is in the small intestine. There, the partially-digested carbohydrates are degraded again by the pancreatic amylase (an enzyme secreted by the pancreas). The enzyme cleaves the glycosidic bond resulting in the degradation of the complex carbohydrates into simple sugars. The brush border of the small intestine releases digestive enzymes such as isomaltase, maltase, sucrase, and lactase. Isomaltase digests polysaccharides at the alpha 1-6 linkages, and convert alpha-limit dextrin to maltose. Maltase breaks down maltose (a disaccharide) into two glucose units. Sucrase and lactase digest sucrose and lactose, respectively, into monosaccharide constituents. The epithelial cells at the brush border of the small intestine absorb the monosaccharides, and later release them into the bloodstream to be taken up by the different cells of the body. The liver cells take glucose for use in various metabolic activities and store the excess as glycogen.
Biological reactions
Glycogen degradation may occur either in the cytosol or inside the lysosome. In the cytosol, glycogen is broken down by the action of the enzymes glycogen phosphorylase and glycogen debranching enzyme. Glycogen phosphorylase catalyzes the release of glucose 1-phosphate from a linear glycogen chain by cleaving α-(1,4) glycosidic bond. In the presence of limit dextrin ( i.e. a polysaccharide fragment from the glycogen branch point), a different enzyme is needed to complete glycogen degradation. Glycogen debranching enzyme “unties” the glycosidic linkage at branch points to release free glucose. Glucose 1-phosphate in the cytosol may be isomerized into glucose 6-phosphate. The latter is then dephosphorylated to produce free glucose that can be transported outside the cell into the bloodstream.3 In the lysosome, glycogen is degraded through the lysosomal enzyme acid α-glucosidase or acid maltase.3
Biological reactions
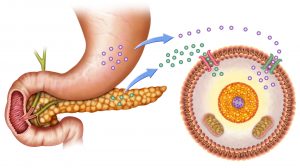
Insulin plays a role in glucose uptake in insulin-stimulated cells, such as those of adipose and muscle tissues. Insulin is released into the bloodstream when the blood glucose level is high. Insulin binds with the insulin receptor in the cell surface. The binding results in the recruitment of certain glucose transporters (GluTs). GluTs facilitate the entry of glucose into the cell. Liver and brain cells are insulin-independent cells, meaning glucose can enter these cells without the prior stimulation of insulin. Liver cells may not require insulin for glucose uptake but insulin still has an effect on them. Insulin activates the enzyme hexokinase that phosphorylates glucose in order to trap it within the cell. It also activates certain enzymes involved in glycogen synthesis, e.g. phosphofructokinase and glycogen synthase. Thus, insulin tells the liver to convert glucose into glycogen by glycogenesis.4
Glucagon is another hormone released into the bloodstream. The pancreas releases glucagon when blood glucose level turns low. This hormone acts by increasing the amount of glucose in the blood. It does so by activating the enzymes involved in glycogenolysis (and gluconeogenesis) in the liver. It tells the hepatocytes to depolymerize glycogen to release glucose.
Biological functions
Although fatty acids are much more energy-rich than glycogen, glycogen remains to be the preferred form of energy storage compounds in animals. The excess glucose is stored in glycogen granules especially in the cells of the liver, muscle, and adipose tissues. Glycogen is non-osmotic whereas glucose is osmotic. Thus, if the excess glucose is not stored as glycogen, it can cause disruption in the osmotic pressure, and eventually cause cell damage or cell death.
Glycogen is an accessible source of glucose. In the muscle and fat cells, glycogen provides them glucose that they can metabolize locally. Since these cells lack the enzyme glucose 6-phosphatase, the glucose is used internally and is not shared with other cells. In contrast, the liver cell has glucose 6-phosphatase that can dephosphorylate the trapped glucose, and thereby, allows glucose to be mobilized out of the liver cell. When there is not enough glucose circulating in the bloodstream, the pancreas secretes glucagon that stimulates the liver cells to undergo glycogenolysis and release free glucose into the bloodstream. Hence, glycogen helps in maintaining normal blood glucose levels. Similar to a “bank”, the body can “save up” the extra glucose and then “withdraw” glucose when there is energy demand. Glucose is an essential fuel. It is the chief energy source preferred by the brain. Furthermore, glucose, unlike fatty acids, can supply energy even during an anaerobic (oxygen-deprived) activity.2
Inborn errors of metabolism
Glycogenosis is the condition characterized by the inability of the body to metabolize glycogen properly. There are different types of glycogenosis depending on the enzyme deficiency involved. Each of these types involves a different dysfunctional gene, and therefore a different enzyme deficiency. These types are as follows:
- Type 0 glycogenosis
- Type 1 glycogenosis (von Gierke’s disease)
- Type 2 glycogenosis (Pompe disease)
- Type 3 glycogenosis (Cori’s disease or Forbes’ disease)
- Type 4 glycogenosis (Andersen disease)
- Type 5 glycogenosis (McArdle disease)
- Type 6 glycogenosis (Hers’ disease)
- Type 7 glycogenosis (Tarui’s disease)
- Type 9 glycogenosis
- Type 10 glycogenosis
- Type 12 glycogenosis (aldolase A deficiency)
Supplementary
Etymology
- French glycogène, from Ancient Greek γλυκύς (“glukús”, meaning “sweet”) + -γενής (meaning “–genḗs”)
Synonym(s)
Derived term(s)
- Brancher glycogen storage disease
- Glycogen debranching enzyme system
- Glycogen storage disease
- Glycogen storage disease type I
- Glycogen storage disease type II
- Glycogen storage disease type IIb
- Glycogen storage disease type IV
- Glycogen storage disease type IX
- Glycogen synthetase deficiency
Further reading
Compare
- (plant) starch
See also
Reference
- Young, F. G. (1957). “Claude Bernard and the Discovery of Glycogen”. British Medical Journal. 1(5033 (Jun. 22, 1957)): 1431–7. doi:10.1136/bmj.1.5033.1431 ://www.bmj.com/content/1/5033/1431 Link
- Berg, J. M., Tymoczko, J. L., & Lubert Stryer. (2002, January 1). Glycogen Metabolism. Retrieved from ://www.ncbi.nlm.nih.gov/books/NBK21190/ Link
- Adeva-Andany, M. M., González-Lucán, M., Donapetry-García, C., Fernández-Fernández, C., & Ameneiros-Rodríguez, E. (2016). Glycogen metabolism in humans. BBA Clinical, 5, 85–100. ://doi.org/10.1016/j.bbacli.2016.02.001 Link
- Physiologic Effects of Insulin. (2019, January 1). Retrieved from ://www.vivo.colostate.edu/hbooks/pathphys/endocrine/pancreas/insulin-phys.html Link
© Biology Online. Content provided and moderated by Biology Online Editors