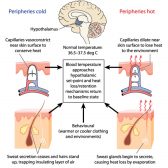
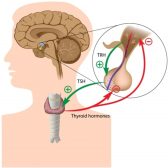
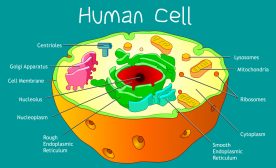
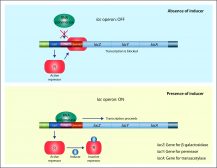
Definition
noun
A type of mucopolysaccharides and mucolipids
Supplement
Lysosomal storage disease is a collective term for the various metabolic disorders due to defects in lysosomal function resulting in an abnormal accumulation of toxic materials in the cell. One of them is mucolipidosis, a hereditary metabolic disorder caused by a deficiency in the enzyme N-acetylglucosamine-1-phosphotransferase. This enzyme takes part in the metabolism of mucopolysaccharides and mucolipids. The absence of this enzyme or the insufficiency of the production of this enzyme leads to the accumulation of mucopolysaccharides and mucolipids in various tissues. Because of this, it leads to the manifestation of the symptoms characterizing mucolipidosis.
This condition is hereditary and is inherited in autosomal recessive pattern. This means that the child with mucolipidosis has two copies of the defective gene that codes for N-acetylglucosamine-1-phosphotransferase. This enzyme is important in the phosphorylation of carbohydrate residues on N-linked glycoproteins.
There are four types of mucolipidosis: type I (also called sialidosis), type II (also called inclusion-cell disease), type III (also referred to as pseudo-Hurler polydystrophy), and type IV (also called mucolipidin 1 deficiency). Type I, though, is now categorized under glycoproteinosis whereas type IV is now regarded as a type of gangliosidosis.
Abbreviation/Acronym:
- ML
See also: